Soft robotic nerve cuffs could revolutionize treatment of neurological conditions
Posted by Kostas Pantremenos in REHABILITATION, Rehabilitation robotics on May 4, 2024
Soft robotic nerve cuffs could revolutionize treatment of neurological conditions
Source: Soft robotic nerve cuffs could revolutionize treatment of neurological conditions
[Webinar] Join IBIA’s Webinar Event
Posted by Kostas Pantremenos in Fatigue, Radiology/Imaging technology on May 1, 2024

Join IBIA’s Webinar Event
Join your colleagues from around the world for a wide variety of learning opportunities through December 2024. Registration is free for IBIA Members and $25 for non-members. We look forward to welcoming you to the 2024 IBIA Webinar Series. Questions: congress@internationalbrain.org
Registration links are below.
Post Traumatic Fatigue: Best Practices Assessment and Clinical Management
Speaker: Brian Greenwald, MD
Date and Time: May 9, 2024 at 12 PM ET
Organized by North America Brain Injury Society.
This webinar will review the definition of fatigue as well as how it is conceptualized including peripheral versus central fatigue. Anatomic areas of the brain implicated in fatigue will be reviewed. Assessment tool for fatigue, the differential diagnosis, medical work up and a range of treatment considerations will be discussed.
Brian Greenwald, MD, is the Medical Director of the Center for Brain Injuries and the Associate Medical Director of JFK Johnson Rehabilitation Institute. He is the Fellowship Director for JFK Johnson’s Accreditation Council for Graduate Medical Education (ACGME)- certified Brain Injury Medicine fellowship. Additionally, Dr. Greenwald is a Clinical Associate Professor in the Department of Physical Medicine and Rehabilitation at Rutgers Robert Wood Johnson Medical School and is a Core Associate Professor in the Department of Physical Medicine and Rehabilitation at the Hackensack Meridian School of Medicine.

Registration – Post Traumatic Fatigue Webinar
Functional Brain Imaging in DoC Clinical Practice and the Forensic Arena: What Can Be Said with Confidence
Speaker: Steven Laureys, MD, PhD, FEAN
Date and Time: May 29, 2024 at 11 AM ET
Organized by Disorders of Consciousness Special Interest Group.
This talk will explore the role of functional brain imaging in assessing patients with disorders of consciousness (DoC) both in clinical practice and forensic settings. We will discuss the current state of knowledge regarding the use of functional imaging techniques such as functional magnetic resonance imaging (fMRI) and positron emission tomography (PET), highlighting what can be confidently inferred from these methods in terms of diagnosis, prognosis, and the detection of covert consciousness. The talk will also address the challenges and limitations of these techniques, emphasizing the need for cautious interpretation and the integration of multiple assessment tools in comprehensive patient care.

Dr. Steven Laureys is a prominent neurologist and neuroscientist known for his work in the field of consciousness research. He headed, until early 2020, the Coma Science Group that he created in 2014 within the GIGA Consciousness Centre at the University of Liège. Most of his research work is devoted to the study of alterations in consciousness in severely brain-damaged patients, as well as during anesthesia, sleep, meditation and in the hypnotic state. Steven Laureys is the author of over 450 scientific articles, some of which have been published in the most prestigious journals. Winner of numerous scientific awards, including the Francqui Prize (2017) – Belgium’s most prestigious scientific prize – and the Generet Prize (2019), Steven Laureys is undoubtedly one of the world’s leading specialists on the issue of altered states. He is currently professor at the CERVO research centre at Laval University (Quebec, QC, Canada).
Registration – Functional Brain Imaging
For more information on the IBIA webinars, please contact congress@internationalbrain.org.
[Abstract] Evaluation of custom-made VR exergame for at-home Stroke rehabilitation. A longitudinal single-arm study. – Full Text PDF
Posted by Kostas Pantremenos in Paretic Hand, Video Games/Exergames, Virtual reality rehabilitation on April 26, 2024
Abstract
Exercise games (Exergames) based on Virtual Reality (VR) have emerged as a promising option for supporting physical rehabilitation in stroke users. As a com- plementary therapy, they offer valuable benefits such as therapy engagement and enjoyment. In this study, we assessed the effectiveness of an immersive, custom- made VR exergame designed for upper limb rehabilitation in stroke participants aged 50 and above. We conducted 14 sessions of 15 minutes involving ten par- ticipants (6 females, ages 58.1 ± 7.5 years old) who volunteered to participate in an assisted at-home rehabilitation process. The study employed a range of evaluation tests to measure physical rehabilitation and game user experience out- comes. The tests included pre- and post-assessments of range of motion (ROM), the Ashworth spasticity test, and the Borg rating of perceived fatigue question- naire. To evaluate the game participant experience, we used the VR Neuroscience Questionnaire (VRNQ), and the Immersive Tendencies Questionnaire (ITQ). Our results revealed significant improvements in the range of motion for elbow and shoulder flexion, extension, adduction, and abduction. Furthermore, we observed a reduction in Ashworth spasticity, and the fatigue scale showed reduced per- ception comparing the last with the first session, although the difference was insignificant. The VRNQ questionnaire indicated significant enhancements in the domains related to ”Game Experience” and ”Game Mechanics” and an overall reduction of the perceived “Motion Sickness”. In the ITQ questionnaire, partic- ipants reported high levels of ”Attention,” and while there were no significant differences in ”Immersion” and ”Enjoyment,” a considerable improvement was observed in ”Excitement”. In summary, our results indicate that the immersive VR exergame improved the range of motion, spasticity, and overall game user experience among participants with stroke in a longitudinal, single-arm inter- vention. We conclude that using custom-made VR exergames is an effective and motivating tool for upper limb rehabilitation, with positive changes in both clin- ical and perception outcomes, and the positive and measurable effects persist after the first sessions. These findings support using VR exergames as a comple- mentary tool for at-home rehabilitation therapy with good ease of use, improved physical rehabilitation outcomes, and high treatment adherence.
[Abstract] Hybrid Brain-Computer Interface Controlled Soft Robotic Glove for Stroke Rehabilitation
Posted by Kostas Pantremenos in Paretic Hand, Rehabilitation robotics on April 26, 2024
Abstract:
Soft robotic glove controlled by a brain-computer interface (BCI) have demonstrated effectiveness in hand rehabilitation for stroke patients. Current systems mostly rely on static visual representations for patients to perform motor imagination (MI) tasks, resulting in lower BCI performance. Therefore, this study innovatively used MI and high-frequency steady-state visual evoked potential (SSVEP) to construct a friendly and natural hybrid BCI paradigm. Specifically, the stimulation interface sequentially presented decomposed action pictures of the left and right hands gripping a ball, with the pictures flashing at specific stimulation frequencies (left: 34 Hz, right: 35 Hz). Integrating soft robotic glove as feedback, we established a comprehensive “peripheral – central – peripheral” hand rehabilitation system to facilitate the hand rehabilitation of patients. Filter bank common spatial pattern (FBCSP) and filter bank canonical correlation analysis (FBCCA) algorithms were used to identify MI and SSVEP signals, respectively. Additionally, to fuse the features of these two signals, we proposed a novel fusion algorithm for improving the recognition accuracy of the system. The feasibility of the proposed system was validated through online experiments involving 12 healthy subjects and 9 stroke patients, achieving accuracy rates of 95.83 ± 6.83% and 63.33 ± 10.38%, respectively. The accuracy of MI and SSVEP in 12 healthy subjects reached 81.67 ± 15.63% and 95.14 ± 7.47%, both lower than the accuracy after fusion, these results confirmed the effectiveness of the proposed algorithm. The accuracy rate was more than 50% in both healthy subjects and patients, confirming the effectiveness of the proposed system.
[WEB] How common is Epilepsy?
Posted by Kostas Pantremenos in Epilepsy on April 23, 2024
Epilepsy – An Overview

Epilepsy is not one single condition. Rather, it is defined as a spectrum of disorders that involve abnormal activity within the brain. “Epilepsy” means the same thing as the term “seizure disorders.” In epilepsy, abnormal brain waves disturb electrical activity, leading to seizures. Symptoms of epileptic seizures include having unusual sensations or emotions, behaving in unusual ways, or experiencing convulsions or loss of consciousness. Brain damage, illness, and irregular brain development can all cause abnormal brain waves that lead to seizures.
Approximately half of all people who have had one seizure will have more (typically within six months). However, in order to be diagnosed with epilepsy, a person must have had more than one seizure, and doctors must consider it likely that they will continue to have seizures. When a person has a seizure that lasts for more than 5 minutes, or a person has more than one seizure within a 5 minutes period (without returning to consciousness between the seizures), it is called status epilepticus.
Some forms of epilepsy last for a limited time, although the condition is often lifelong. Although there is no cure currently for epilepsy, there are many treatments available for the condition. About 70 percent of people with seizures can control them with proper diagnosis and use of medication.
There are many different types of seizures, and some people with epilepsy will experience more than one type. Some examples of seizure types include:
- Absence seizures
- Generalized tonic-clonic seizures
- Atonic (or akinetic) seizures
The History of Epilepsy
People have been aware of epilepsy and seizures for millennia. A Babylonian medical textbook made up of 40 tablets and dating to 2000 B.C. contains a chapter that accurately describes many of the different types of seizures known today. However, seizures were thought to be supernatural in cause — each seizure type was associated with a different evil spirit or angry god — so the treatments prescribed were spiritual.
Epilepsy in Ancient Greece
The word “epilepsy” comes from the Greek word “epilepsia,” meaning “to seize” or “to take hold of.” By the 5th century B.C., the Greeks still considered epilepsy a “sacred” disease. Yet the renowned ancient physician Hippocrates described epilepsy as a brain disorder. This was a radical idea for the time. He recommended physical treatments while also recognizing that if the seizures became chronic, the disorder was incurable.
Despite Hippocrates’ writings, epilepsy continued to be considered a supernatural condition for the next two millennia. People with epilepsy were subjects of immense social stigma, treated as outcasts, and even punished as witches. In many places, people who suffered seizures were prevented from going to school, working, marrying, and having children. There were a few people with prominent positions thought to have had epilepsy — including Julius Caesar, Tsar Peter the Great of Russia, Pope Pius IX, and Fyodor Dostoevsky — but most people with epilepsy were prevented from living as full members of society.
Epilepsy in the 14th Century and Beyond
During the Renaissance, some scientists tried to prove epilepsy was a physical, not spiritual, illness. Then, in the 19th century, neurology became a recognized medical discipline and the idea of epilepsy as a brain disorder became normal in North America and Europe. In 1857, Sir Charles Lacock introduced bromide of potassium as the first antiepileptic drug (AED).
In 1873, a British neurologist named John Hughlings Jackson first described epilepsy as we understand it today. Jackson showed that seizures are caused by sudden, brief electrochemical discharges of energy in the brain. In 1909, the International League Against Epilepsy was founded as a global professional organization of epileptologists.
By the 1920s, Hans Berger, a German psychiatrist, had developed the electroencephalogram (EEG) to measure brain waves. It showed that each type of seizure is associated with a different brain wave pattern. The EEG also aided in the discovery that specific sites in the brain were responsible for seizures and expanded the potential for surgical treatments. Surgery became a more widely available option by the 1950s.
The Development of Antiepileptic Drugs
The medication phenobarbital was identified as an AED in 1912, and phenytoin (sold under the brand names Dilantin and Phenytek) was developed in 1938. Carbamazepine (sold under the brand names Tegretol and Carbatrol) was identified in 1953. These drugs have since been approved by the U.S. Food and Administration (FDA) and continue to be used today.
An accelerated drug-discovery process began in the 1970s with the creation of the Anticonvulsant Screening Program, sponsored by the National Institute of Neurological Disorders and Stroke. The program helped scientists gain a better understanding of the brain and epilepsy. Scientists have strived to reduce serious side effects associated with the use of older AEDs through drug-development processes.
Keppra (levetiracetam) was approved by the FDA in 1999. Several newer drugs, including Vimpat (lacosamide), Briviact (brivaracetam), and Aptiom (eslicarbazepine acetate), have been introduced in the past 10 to 15 years. Other promising medications are also in the pipeline.
Social Stigma
The stigma around epilepsy has lessened as more people are able to effectively treat their seizures. However, epilepsy largely remains an “invisible” illness. Millions of people in developing countries do not have access to AEDs, and stigma and discrimination are still widespread, especially in places where people still believe that seizures have a supernatural cause.
How Common Is Epilepsy?
People of all backgrounds, races, ethnicities, and ages are equally affected by epilepsy. It is estimated that epilepsy affects 1.2 percent of the population of the United States and more than 50 million people worldwide, making it one of the most common neurological disorders. Approximately 45,000 children under the age of 18 are diagnosed with epilepsy every year in the U.S., and roughly 10.5 million children worldwide live with epilepsy.
Diagnosing Epilepsy
Neuroimaging capabilities have improved over the past few decades. Magnetic resonance imaging (MRI), computerized tomography (CT) scans, and other techniques are able to detect more and more subtle brain lesions responsible for epilepsy.
Read more about diagnosing epilepsy.
Causes of Epilepsy
Causes of epilepsy include a wide variety of brain-related issues, such as structural damage, infectious diseases (like encephalitis), and genetic anomalies. Risk factors for epilepsy include:
- Age
- A family history of seizures
- Head injuries (such as traumatic brain injury or TBI)
- A history of seizures as a child
Read more about causes of seizures and epilepsy.
Types of Seizures
There are many types of epilepsy. Seizures are broken into two categories: focal seizures and generalized seizures. Focal seizures can be categorized by whether or not there is a loss of consciousness or awareness. Generalized seizures can be further broken down into absence, tonic, atonic, clonic, myoclonic, and tonic-clonic seizures.
Learn more about seizure types and symptoms of epilepsy.
Condition Guide
[Abstract] Evaluation of custom-made VR exergame for at-home Stroke rehabilitation. A longitudinal single-arm study. – Full Text PDF
Posted by Kostas Pantremenos in Tele/Home Rehabilitation, Video Games/Exergames, Virtual reality rehabilitation on April 21, 2024
Abstract
Exercise games (Exergames) based on Virtual Reality (VR) have emerged as a promising option for supporting physical rehabilitation in stroke users. As a com- plementary therapy, they offer valuable benefits such as therapy engagement and enjoyment. In this study, we assessed the effectiveness of an immersive, custom- made VR exergame designed for upper limb rehabilitation in stroke participants aged 50 and above. We conducted 14 sessions of 15 minutes involving ten par- ticipants (6 females, ages 58.1 ± 7.5 years old) who volunteered to participate in an assisted at-home rehabilitation process. The study employed a range of evaluation tests to measure physical rehabilitation and game user experience out- comes. The tests included pre- and post-assessments of range of motion (ROM), the Ashworth spasticity test, and the Borg rating of perceived fatigue question- naire. To evaluate the game participant experience, we used the VR Neuroscience Questionnaire (VRNQ), and the Immersive Tendencies Questionnaire (ITQ). Our results revealed significant improvements in the range of motion for elbow and shoulder flexion, extension, adduction, and abduction. Furthermore, we observed a reduction in Ashworth spasticity, and the fatigue scale showed reduced per- ception comparing the last with the first session, although the difference was insignificant. The VRNQ questionnaire indicated significant enhancements in the domains related to ”Game Experience” and ”Game Mechanics” and an overall reduction of the perceived “Motion Sickness”. In the ITQ questionnaire, partic- ipants reported high levels of ”Attention,” and while there were no significant differences in ”Immersion” and ”Enjoyment,” a considerable improvement was observed in ”Excitement”. In summary, our results indicate that the immersive VR exergame improved the range of motion, spasticity, and overall game user experience among participants with stroke in a longitudinal, single-arm inter- vention. We conclude that using custom-made VR exergames is an effective and motivating tool for upper limb rehabilitation, with positive changes in both clin- ical and perception outcomes, and the positive and measurable effects persist after the first sessions. These findings support using VR exergames as a comple- mentary tool for at-home rehabilitation therapy with good ease of use, improved physical rehabilitation outcomes, and high treatment adherence.
[Abstract] Impact of telehealth on stroke survivor–caregiver dyad in at-home rehabilitation: A systematic review
Posted by Kostas Pantremenos in Caregivers, Tele/Home Rehabilitation on April 19, 2024
Abstract
Aim
To examine studies involving the impact of telerehabilitation (TLR), tele-training and tele-support on the dyad stroke survivor and caregiver in relation to psychological, physical, social and health dimensions.
Design
A systematic review was conducted.
Data Sources
The following electronic databases were consulted until September 2023: PsycInfo, CINAHL, Eric, Ovid, PubMed, Scopus, Cochrane Central and Web of Science.
Review Methods
It was conducted and reported following the checklists for Reviews of PRISMA 2020 Checklist. Critical evaluation of the quality of the studies included in the review was performed with the Joanna Briggs Institute Checklists.
Data Synthesis
A total of 2290 records were identified after removing duplicates, 501 articles were selected by title and abstract and only 21 met the inclusion criteria. It included 4 quasi-experimental studies, 7 RCTs, 1 cohort study and 9 qualitative studies. The total number of participants between caregivers and stroke survivors was 1697, including 858 stroke survivors and 839 caregivers recruited from 2002 to 2022. For a total of 884 participants who carried out TLR activities in the experimental groups,11 impact domains were identified: cognitive/functional, psychological, caregiver burden, social, general health and self-efficacy, family function, quality of life, healthcare utilization, preparedness, quality of care and relationship with technology.
Conclusions
The results support the application of telehealth in the discharge phase of hospitals and rehabilitation centres for stroke survivors and caregivers. TLR could be considered a substitute for traditional rehabilitation only if it is supported by a tele-learning programme for the caregiver and ongoing technical, computer and health support to satisfy the dyad’s needs.
[ARTICLE] Noninvasive spinal stimulation improves walking in chronic stroke survivors: a proof-of-concept case series – Full Text
Posted by Kostas Pantremenos in Gait Rehabilitation - Foot Drop on April 8, 2024
Abstract
Background
After stroke, restoring safe, independent, and efficient walking is a top rehabilitation priority. However, in nearly 70% of stroke survivors asymmetrical walking patterns and reduced walking speed persist. This case series study aims to investigate the effectiveness of transcutaneous spinal cord stimulation (tSCS) in enhancing walking ability of persons with chronic stroke.
Methods
Eight participants with hemiparesis after a single, chronic stroke were enrolled. Each participant was assigned to either the Stim group (N = 4, gait training + tSCS) or Control group (N = 4, gait training alone). Each participant in the Stim group was matched to a participant in the Control group based on age, time since stroke, and self-selected gait speed. For the Stim group, tSCS was delivered during gait training via electrodes placed on the skin between the spinous processes of C5–C6, T11–T12, and L1–L2. Both groups received 24 sessions of gait training over 8 weeks with a physical therapist providing verbal cueing for improved gait symmetry. Gait speed (measured from 10 m walk test), endurance (measured from 6 min walk test), spatiotemporal gait symmetries (step length and swing time), as well as the neurophysiological outcomes (muscle synergy, resting motor thresholds via spinal motor evoked responses) were collected without tSCS at baseline, completion, and 3 month follow-up.
Results
All four Stim participants sustained spatiotemporal symmetry improvements at the 3 month follow-up (step length: 17.7%, swing time: 10.1%) compared to the Control group (step length: 1.1%, swing time 3.6%). Additionally, 3 of 4 Stim participants showed increased number of muscle synergies and/or lowered resting motor thresholds compared to the Control group.
Conclusions
This study provides promising preliminary evidence that using tSCS as a therapeutic catalyst to gait training may increase the efficacy of gait rehabilitation in individuals with chronic stroke.
Trial registration NCT03714282 (clinicaltrials.gov), registration date: 2018-10-18.
Background
Stroke is the leading cause of adult-onset disability [1]. Despite many advances in gait research in the last decade, about 35% of stroke survivors fail to regain independence in performing activities of daily living due to the impaired function of their affected leg, and about 70% have gait deficits, including reduced walking speeds, asymmetrical walking patterns, and motor coordination issues [2,3,4].
Walking deficits after stroke mostly derive from a disruption of the corticospinal pathways that play an important role in transmitting sensory–motor commands [5, 6]. To address this, most interventions using non-invasive electrical pulses focus on stimulation of the motor cortex to activate dormant or new pathways [2, 7, 8]. However, while supra-spinal regions can facilitate fine locomotor control, spinal networks ultimately generate the basic locomotor pattern [9, 10]. More interestingly, a recent study using functional MRI showed increased blood-oxygen-level dependent activities in motor cortex following transcutaneous spinal cord stimulation (tSCS) in individuals with stroke [11]. Therefore, we hypothesized that tSCS would facilitate an improvement of gait after stroke. Our previous work, in collaboration with additional researchers, established anatomical and physiological changes in the spinal cord after stroke [12, 13], offering a theoretical basis for testing our hypothesis of targeting the spinal circuits for post-stroke recovery.
Recently, Moshonkina et al. reported functional improvements in post-stroke individuals after 2 weeks of tSCS with standard physical therapy, achieving the minimum clinical important differences (MCID) in the 6 min walk test and comfortable walking speed [14]. The same investigators reported immediate improvements in walking kinematics after a single tSCS session [15, 16]. Notably, however, none of the studies mentioned above investigated the effects of more than 4 weeks of training nor tried to explore the potential neurophysiological differences accompanied with gait outcomes. Consequently, it remains unclear whether tSCS can exert a lasting impact on restoration of function following a stroke.
We investigated whether tSCS combined with symmetry-focused gait training has a sustained effect on gait recovery after chronic stroke. We hypothesized that longer-term gait training (24 sessions) with tSCS would lead to greater sustained improvements in walking function compared to control treatment focused solely on gait training. Specifically, we focused on gait symmetry since such improvements can have lasting effects on balance and overall mobility of stroke survivors [6]. We also expected that gait improvements would be associated with physiological changes in muscle coordination measured from electromyography (EMG) of the paretic side, and spinal excitability determined by the spinal motor evoked responses (sMERs). […]
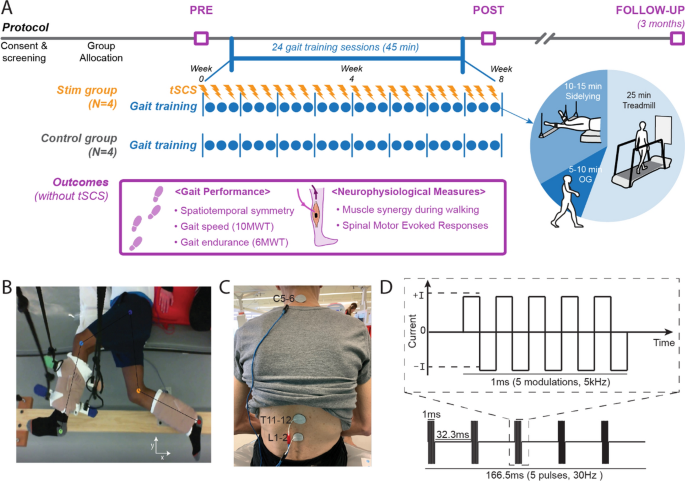
Study protocol and stimulation setup. A Overall experimental protocol. B Top–down view of position of the legs extended beyond the edge of the table and supported with vertically cables during the side-lying training of a participant (Stim 2). C tSCS delivered using surface electrodes on the skin between the C5–6, T11–12, and L1–2 spinous processes (cathode) and a surface electrode on each anterior crest (anode, not shown). D Schematic representation of biphasic pulse sequence used for tSCS. tSCS transcutaneous spinal cord stimulation, OG overground walking, 10MWT 10-m walk test, 6MWT 6-min walk test
[Abstract] Neurostimulation for treatment of post-stroke impairments
Posted by Kostas Pantremenos in Epilepsy on April 8, 2024
Abstract
Neurostimulation, the use of electrical stimulation to modulate the activity of the nervous system, is now commonly used for the treatment of chronic pain, movement disorders and epilepsy. Many neurostimulation techniques have now shown promise for the treatment of physical impairments in people with stroke. In 2021, vagus nerve stimulation was approved by the FDA as an adjunct to intensive rehabilitation therapy for the treatment of chronic upper extremity deficits after ischaemic stroke. In 2024, pharyngeal electrical stimulation was conditionally approved by the UK National Institute for Health and Care Excellence for neurogenic dysphagia in people with stroke who have a tracheostomy. Many other approaches have also been tested in pivotal device trials and a number of approaches are in early-phase study. Typically, neurostimulation techniques aim to increase neuroplasticity in response to training and rehabilitation, although the putative mechanisms of action differ and are not fully understood. Neurostimulation techniques offer a number of practical advantages for use after stroke, such as precise dosing and timing, but can be invasive and costly to implement. This Review focuses on neurostimulation techniques that are now in clinical use or that have reached the stage of pivotal trials and show considerable promise for the treatment of post-stroke impairments.
Key points
- Neurostimulation techniques are ideally suited for use during stroke recovery owing to their ability to target anatomical structures or neuronal networks, alongside precise timing and dosing.
- Paired invasive vagus nerve stimulation has been shown to increase the number of people who achieve clinically important improvements in upper extremity impairment and performance of functional tasks following stroke. The treatment is now in clinical use in the USA.
- Several other neurostimulation techniques show promise for post-stroke impairments but definitive data from adequately powered trials are lacking.
- Pharyngeal electrical stimulation increases the odds of decannulation following tracheostomy and is under investigation as a treatment for post-stroke dysphagia.

