Archive for category Neuroplasticity
[Review] The evolution of neuromodulation for chronic stroke: From neuroplasticity mechanisms to brain-computer interfaces – Full Text
Posted by Kostas Pantremenos in Neuroplasticity, REHABILITATION on March 1, 2024
Abstract
Stroke is one of the most common and debilitating neurological conditions worldwide. Those who survive experience motor, sensory, speech, vision, and/or cognitive deficits that severely limit remaining quality of life. While rehabilitation programs can help improve patients’ symptoms, recovery is often limited, and patients frequently continue to experience impairments in functional status. In this review, invasive neuromodulation techniques to augment the effects of conventional rehabilitation methods are described, including vagus nerve stimulation (VNS), deep brain stimulation (DBS) and brain-computer interfaces (BCIs). In addition, the evidence base for each of these techniques, pivotal trials, and future directions are explored. Finally, emerging technologies such as functional near-infrared spectroscopy (fNIRS) and the shift to artificial intelligence-enabled implants and wearables are examined. While the field of implantable devices for chronic stroke recovery is still in a nascent stage, the data reviewed are suggestive of immense potential for reducing the impact and impairment from this globally prevalent disorder.
Introduction
Due to improving techniques for treating acute stroke, more patients than ever are entering the chronic stroke phase during which motor recovery becomes significantly more challenging [1]. Approximately 34% of the global total healthcare expenditure is spent on stroke, and in the US, the economic burden of chronic stroke increases by approximately $140,000 for treatment, rehabilitation and supportive care over the course of a typical patient’s lifetime [[2], [3], [4]]. Furthermore, incidence rates of chronic stroke are projected to grow due to a global increase in population age [2]. These worrisome trends underline the crucial need for effective rehabilitation to improve quality-of-life and enable patients to recover functional ability post-stroke.
The current standard-of-care for post-stroke recovery is physical rehabilitation, which exploits the innate neuroplasticity of the brain to restore function [1,5]. Physical rehabilitation programs, especially when delivered as soon as possible after the onset of stroke, can be highly efficacious [5]. Notwithstanding, the rate of improvement in functional ability regained through physical rehabilitation tends to peak after a few months post-stroke and eventually tapers; minimal improvement is seen after 12 months and many patients remain considerably disabled. Therefore, a critical need exists for interventions that can either increase the rate of functional recovery during the early post-stroke period or that can produce meaningful functional improvement after 12 months. Given that the nature of post-stroke functional recovery is mediated by neuroplastic changes, interventions that increase or prolong neuroplasticity have been the target of recent investigations.
One such intervention is the application of electromagnetic energy to the brain in the form of neuromodulation, which has been shown to be an effective trigger for neuroplastic processes such as synaptogenesis and functional reorganization [6]. Both invasive and non-invasive modalities exist. Non-invasive modalities such as transcranial direct current stimulation (tDCS) and transcranial magnetic stimulation (TMS), have demonstrated improvement of motor function in post-stroke patients [6]. Similarly, invasive modalities such as vagus nerve stimulation (VNS) and deep brain stimulation (DBS) show great promise in improving rehabilitation in stroke patients suffering from disabling symptoms. Moreover, there has been rapid development of therapeutic neurostimulation in the form of brain-computer interfaces (BCIs), which utilizes real-time analysis of brain states to enable automatic adjustment of stimulation parameters [7]. In this paper, we will discuss the landmark trials, current applications, and future directions of the various modalities of invasive neuromodulation for stroke rehabilitation with an emphasis on VNS, DBS and BCI. […]
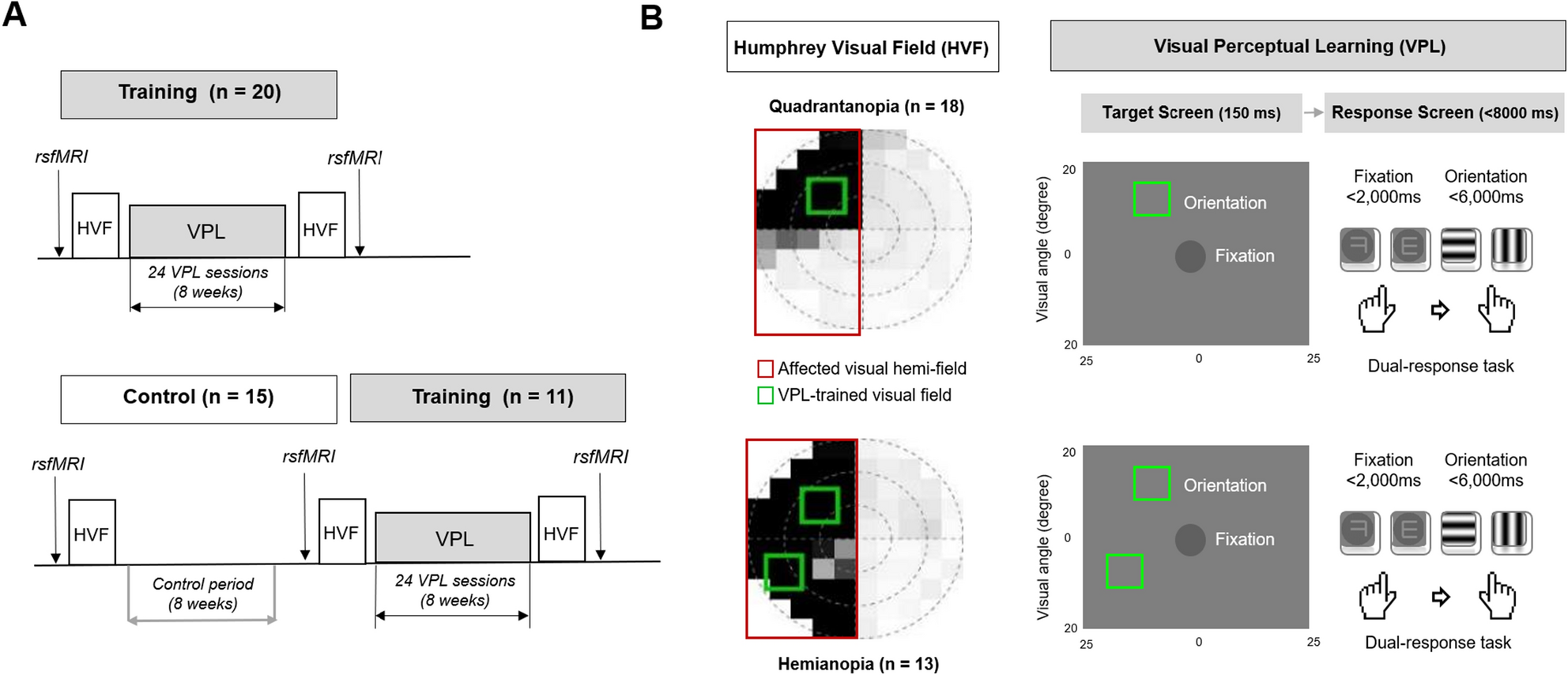
[ARTICLE] Functional connectivity interacts with visual perceptual learning for visual field recovery in chronic stroke – Full Text
Posted by Kostas Pantremenos in Hemianopsia, Neuroplasticity on February 19, 2024
Abstract
A reciprocal relationship between perceptual learning and functional brain changes towards perceptual learning effectiveness has been demonstrated previously; however, the underlying neural correlates remain unclear. Further, visual perceptual learning (VPL) is implicated in visual field defect (VFD) recovery following chronic stroke. We investigated resting-state functional connectivity (RSFC) in the visual cortices associated with mean total deviation (MTD) scores for VPL-induced VFD recovery in chronic stroke. Patients with VFD due to chronic ischemic stroke in the visual cortex received 24 VPL training sessions over 2 months, which is a dual discrimination task of orientation and letters. At baseline and two months later, the RSFC in the ipsilesional, interhemispheric, and contralesional visual cortices and MTD scores in the affected hemi-field were assessed. Interhemispheric visual RSFC at baseline showed the strongest correlation with MTD scores post-2-month VPL training. Notably, only the subgroup with high baseline interhemispheric visual RSFC showed significant VFD improvement following the VPL training. The interactions between the interhemispheric visual RSFC at baseline and VPL led to improvement in MTD scores and largely influenced the degree of VFD recovery. The interhemispheric visual RSFC at baseline could be a promising brain biomarker for the effectiveness of VPL-induced VFD recovery. […]
Figure 1
[Abstract] Vagus Nerve Stimulation Paired With Rehabilitation for Post-Stroke Arm Impairment: One Year Follow-Up of the VNS-REHAB Pivotal Trial
Posted by Kostas Pantremenos in Neuroplasticity, Paretic Hand, REHABILITATION on February 19, 2024
Abstract
Background: Persistent post-stroke impairment of the arm and hand is debilitating after stroke. Pairing vagus nerve stimulation (VNS) with upper extremity (UE) rehabilitation improves such deficits after 5 months and was approved by the FDA in 2021. Here, we present 1-year outcomes from the VNS-REHAB pivotal trial.
Methods: Stroke participants with moderate-to-severe UE impairment were randomized to task-specific rehabilitation plus either active VNS (n=53, VNS) or sham VNS (n=55, Control). After baseline assessment (Pre-therapy), both groups did 6 wk. of in-clinic therapy followed by a 3-mo. home exercise program combined with active or sham VNS (post90). Controls then crossed over to receive 6 wk. of active VNS followed by a 3-mo. home exercise program (Cross-over post90). Both groups continued active VNS with a home exercise program through 1 year, after which change from Pre-therapy baseline in Fugl-Meyer Assessment-Upper Extremity (FMA-UE) and Wolf Motor Function Test (WMFT) scores were obtained. To determine whether participants made additional gains, 1 year outcome scores (n=70) were also compared to post90 (n=38, VNS) and Cross-over post90 (n=32, Control) scores. Data was available from 74 participants at one year, with others not available mainly due to COVID-19.
Results: At 1-year, both FMA-UE and WMFT scores improved from Pre-therapy baseline by 5.3±6.9 (CI=3.7-6.9, p<0.001) and 0.51±0.52 (CI=0.39-0.63, p<0.001) points, respectively. FMA-UE change at 1-year was not significantly different from the post90 (VNS) and Cross-over post90 (control) timepoints (n=70, mean difference: -0.3±4.1, CI=-1.3-0.67, p=0.52), but WMFT was, by an additional 0.09 points (n=70, mean difference: 0.09±0.35, CI=0.01-0.18, p=0.03), indicating that participants either improved or maintained motor gains through 1 year.
Conclusion: Improvements in arm and hand function with VNS were maintained at 1-year follow-up, supporting use of VNS paired with rehabilitation as a long-term treatment option for individuals with post-stroke UE impairment. Limitations include sample size and lack of details of therapeutic regimens over the long term. Future studies and an ongoing clinical registry will explore the long-term impact of active VNS in real-world settings.
[BLOG POST] Stroke Recovery Plateau: Why Progress Slows Down & How to Keep Going
Posted by Kostas Pantremenos in Neuroplasticity, Recovery Plateau on January 14, 2024
Medically reviewed by Mariah Cairer PT, DPT — written by Flint Rehab.

You may hear a lot of different phrases regarding rehabilitation after a stroke, but what is a stroke recovery plateau? During the early stages of stroke rehabilitation, survivors often make rapid gains in function. After a few months, however, progress may slow or stall, which can lead to discouragement or frustration during the rehabilitation journey. Therapists call this slowing of progress a plateau phase.
While this can be a difficult hurdle during the rehab process, a stroke recovery plateau is a common phase that many survivors experience. Contrary to how this slowing of progress may feel, a plateau after stroke does not mean your recovery has ended. It is possible to push through a stroke recovery plateau and continue working toward your goals.
To help you understand more about a stroke recovery plateau, this article will explain the possible causes of a plateau as well as a general timeline for this slowing of progress. Additionally, we will review tips and tricks to help you overcome a stroke recovery plateau if it occurs.
What Is a Stroke Recovery Plateau?
A stroke recovery plateau is a period of little or no gains in function after a period of rapid progress. These plateaus are frustrating and can make you feel as though you will make no further progress toward your rehabilitation goals.
Most commonly, a stroke recovery plateau occurs around 3-6 months post-stroke. This is roughly the same time that spontaneous recovery slows down, too (more on this soon). During a plateau period, survivors may notice that their speech is not improving as quickly as it once was or that their walking seems to stop progressing. Additionally, survivors may even notice regression of post-stroke symptoms.
In the past, it was commonly believed that recovery ended once a survivor hit their first plateau after stroke. As a result, stroke rehab programs would discourage therapy after six months, citing that improvements in function would no longer be made.
Today we know this is not true as recent research has shown that plateaus are not permanent. Stroke recovery can continue much later after a plateau, as demonstrated by studies investigating rehab interventions in chronic cases of stroke. For example, a recently published report described one man who finally recovered movement in his hand 23 years after his stroke.
Therefore, if you experience a plateau after your stroke, do not give up. Survivors can experience improvement after a stroke recovery plateau, even later than 6 months post-stroke. To help you understand why a stall in progress can occur, we will review the causes of a stroke recovery plateau in the next section.
Causes of Plateaus After Stroke
Before you can learn how to move forward from a stroke recovery plateau, it helps to understand what causes this slowing of progress to begin with. To answer this question, let’s review neuroplasticity; the brain’s natural repair mechanism.
After a stroke, the brain enters a heightened state of plasticity as it heals from injury. This means that it becomes easier for the brain to reorganize itself and recover lost function. When an area of the brain is affected by stroke, neuroplasticity can help rewire functions lost in that area of the brain to other healthy areas.
Heightened plasticity is responsible for most of the spontaneous recovery that occurs after a stroke. It also plays a role in the functional recovery that takes place with consistent rehabilitation post-stroke. Unfortunately, this heightened state of plasticity cannot last forever will and eventually slow.
When neuroplasticity in the brain slows, physical progress will also slow down. This decreased plasticity and slowing of progress is what contributes to a stroke recovery plateau. However, even though neuroplasticity is at a lower level, it is still present. This means you can still make progress, but it may take more work and time.
How to Get Past Stroke Recovery Plateaus
High repetition of task-specific exercise engages the brain’s neuroplasticity, even during a stroke recovery plateau. Therefore, the best way to push through a plateau is to continue with an intentional and consistent therapy regimen. It is important to work closely with your therapy team to develop a plan that addresses your specific secondary effects after stroke and helps you reach your unique goals.
There is a wide variety of exercises for stroke recovery available, but the most important factor in a good recovery program is consistency. However, this can be hard to maintain when it feels like you are not making progress. To help with this, the following are a few ways to boost your motivation and push through the plateau:
1. Add Variety to Your Regimen
Creating a daily routine is a valuable piece of the stroke recovery process. However, a stroke recovery plateau can make your routine feel as if it is no longer as effective as it should be.
While it is important to continue with your daily therapy regimen, changing up your routine can help break up the monotony and increase your motivation level. Some examples of how to add variety to your therapy regimen during a stroke recovery plateau include:
- Trying different exercises. It is important to find exercises that are engaging and fun to help you reach your goals after stroke. For example, try adding in some new balance exercises or tai chi to your home exercise program. Or, if it is safe for you, take your exercise routine outside for a change of scenery. Your physical or occupational therapist can also be a great resource for finding new exercises.
- Finding a hobby. Pick a new skill you want to learn, such as gardening, painting, or learning a new game, and substitute that for an activity you are feeling burnt out on. Using your mental and physical skills for fun hobbies or projects after stroke can help you improve in ways you might not expect. For example, learning a new board game can improve your fine motor function and sharpen your cognitive skills.
- Setting a new goal. Use the acronym SMART to set new goals for stroke recovery. The goal should be Specific, Measurable, Achievable, Relevant, and Time-bound. For example, maybe you will want to increase the amount of time you can stand unsupported from 30 seconds to one minute. Setting new goals can help you break out of a stroke recovery plateau by showing that you are still progressing, even if this progress is slower than it used to be.
These simple changes can add the variety your brain craves during rehabilitation and give you a boost of motivation during a stroke recovery plateau. Talk with your family, friends, and therapy team to come up with more ideas to add variety to your daily routine.
2. Seek Accountability
Another good way to push through a stroke recovery plateau is to find an accountability partner. This could be your physical therapist, a family member, or a close friend. A stroke can cause major changes to daily life and the recovery process is a long journey for most survivors. Finding someone to help you stay motivated as you navigate life after stroke can make a huge difference in your recovery.
For instance, if you experience memory difficulties after stroke, your accountability partner could check in to remind you to do your exercises every day. Or perhaps a friend would like to join you for your exercises or take you to a new fitness class. Having someone to support you and encourage you to exercise can help you overcome this plateau.
3. Try Interactive Home Therapy Equipment
Interactive home therapy devices such as FitMi can be a great addition to your rehabilitation routine and can help you push through a stroke recovery plateau. FitMi combines elements of gaming with therapist-approved stroke recovery exercises, motivating you to perform the high repetition of exercise needed to see improvement in function.
This helps turn therapy into an immersive experience and can prevent boredom during exercise sessions. As a result, survivors can accomplish hundreds of repetitions per half-hour session, which helps to maximize neuroplasticity and push through a recovery plateau after stroke.
Overcoming the Stroke Recovery Plateau
While a stroke recovery plateau is a common aspect of the recovery journey, this can be frustrating for survivors as they see a slowing of functional improvement. However, we know that this stall in progress is likely temporary and that survivors can continue to regain function even decades after their stroke.
Overcoming a recovery plateau is difficult, but not impossible. Discovering new hobbies, finding an accountability partner, and adding variety into your exercise routine are all great ways to help you boost your progress. Most importantly, staying consistent with your stroke recovery exercises is the best way to ensure that you continue progressing toward your goals.
[VIDEO] Post Stroke Foot Dorsiflexion: Using Electrical Stimulation to Reduce Tone & Promote Plasticity – YouTube
Posted by Kostas Pantremenos in Gait Rehabilitation - Foot Drop, Neuroplasticity, Video on December 22, 2023
Further reading on electrophysiology and muscle contractions: https://strokesciences.com/post-strok…
StrokeSciences.Com
[WEB] Exercise Releases Chemical Signals that Boost Brain Health
Posted by Kostas Pantremenos in Neuroplasticity on December 17, 2023

Physical activity is frequently cited as a means of improving physical and mental health. Researchers at the Beckman Institute for Advanced Science and Technology have shown that exercise may also improve brain health more directly. They studied how the chemical signals released by exercising muscles promote neuronal development in the brain.
Their work appears in the journal Neuroscience.
When muscles contract during exercise, like a bicep working to lift a heavy weight, they release a variety of compounds into the bloodstream. These compounds can travel to different parts of the body, including the brain. The researchers were particularly interested in how exercise could benefit a particular part of the brain called the hippocampus.
“The hippocampus is a crucial area for learning and memory, and therefore cognitive health,” said Ki Yun Lee, a PhD student in mechanical science and engineering at the University of Illinois Urbana-Champaign and the study’s lead author. Understanding how exercise benefits the hippocampus could therefore lead to exercise-based treatments for a variety of conditions including Alzheimer’s disease.
To isolate the chemicals released by contracting muscles and test them on hippocampal neurons, the researchers collected small muscle cell samples from mice and grew them in cell culture dishes in the lab. When the muscle cells matured, they began to contract on their own, releasing their chemical signals into the cell culture.
The research team added the culture, which now contained the chemical signals from the mature muscle cells, to another culture containing hippocampal neurons and other support cells known as astrocytes. Using several measures, including immunofluorescent and calcium imaging to track cell growth and multi-electrode arrays to record neuronal electrical activity, they examined how exposure to these chemical signals affected the hippocampal cells.
The results were striking. Exposure to the chemical signals from contracting muscle cells caused hippocampal neurons to generate larger and more frequent electrical signals — a sign of robust growth and health. Within a few days, the neurons started firing these electrical signals more synchronously, suggesting that the neurons were forming a more mature network together and mimicking the organization of neurons in the brain.
However, the researchers still had questions about how these chemical signals led to growth and development of hippocampal neurons. To uncover more of the pathway linking exercise to better brain health, they next focused on the role of astrocytes in mediating this relationship.
“Astrocytes are the first responders in the brain before the compounds from muscles reach the neurons,” Lee says. Perhaps, then, they played a role in helping neurons respond to these signals.
The researchers found that removing astrocytes from the cell cultures caused the neurons to fire even more electrical signals, suggesting that without the astrocytes, the neurons continued to grow — perhaps to a point where they might become unmanageable.
“Astrocytes play a critical role in mediating the effects of exercise,” Lee says. “By regulating neuronal activity and preventing hyperexcitability of neurons, astrocytes contribute to the balance necessary for optimal brain function.”
Understanding the chemical pathway between muscle contraction and the growth and regulation of hippocampal neurons is just the first step in understanding how exercise helps improve brain health.
“Ultimately, our research may contribute to the development of more effective exercise regimens for cognitive disorders such as Alzheimer’s disease,” Lee says
[ARTICLE] Validating the Safe and Effective Use of a Neurorehabilitation System (InTandem) to Improve Walking in the Chronic Stroke Population: Usability Study. – Full Text
Posted by Kostas Pantremenos in Gait Rehabilitation - Foot Drop, Neuroplasticity on November 27, 2023
Abstract
Background:Persistent walking impairment following a stroke is common. Although rehabilitative interventions exist, few exist for use at home in the chronic phase of stroke recovery. InTandem (MedRhythms, Inc) is a neurorehabilitation system intended to improve walking and community ambulation in adults with chronic stroke walking impairment.
Objective:Using design best practices and human factors engineering principles, the research presented here was conducted to validate the safe and effective use of InTandem.
Methods:In total, 15 participants in the chronic phase of stroke recovery (≥6 months after stroke) participated in this validation study. Participants were scored on 8 simulated use tasks, 4 knowledge assessments, and 7 comprehension assessments in a simulated home environment. The number and types of use errors, close calls, and operational difficulties were evaluated. Analyses of task performances, participant behaviors, and follow-up interviews were conducted to determine the root cause of use errors and difficulties.
Results:During this validation study, 93% (14/15) of participants were able to successfully complete the critical tasks associated with the simulated use of the InTandem system. Following simulated use task assessments, participants’ knowledge and comprehension of the instructions for use and key safety information were evaluated. Overall, participants were able to find and correctly interpret information in the materials in order to answer the knowledge assessment questions. During the comprehension assessment, participants understood warning statements associated with critical tasks presented in the instructions for use. Across the entire study, 3 “use errors” and 1 “success with difficulty” were recorded. No adverse events, including slips, trips, or falls, occurred in this study.
Conclusions:In this validation study, people in the chronic phase of stroke recovery were able to safely and effectively use InTandem in the intended use environment. This validation study contributes to the overall understanding of residual use–related risks of InTandem in consideration of the established benefits.
Introduction
Stroke is a major cause of disability and the second leading cause of death worldwide, with its incidence and prevalence expected to increase due to an aging population [1,2]. Many people in the chronic phase of stroke recovery (commonly defined as ≥6 months after stroke) experience walking impairment [3] and consider the ability to walk in their community as “either essential or very important” [4]. Walking rehabilitation can positively impact the well-being of stroke survivors and their families. It can also restore independence—a prospective study in the chronic stroke population reported that better walking ability was positively correlated with quality of life and the ability to live independently [5]. Clinical practice guidelines recommend various interventions for walking impairment, including physical therapy and braces such as an ankle foot orthosis [6–8]. Rhythmic auditory stimulation (RAS) is another clinically effective intervention for the rehabilitation of movements that are naturally rhythmic (eg, walking) [9]. RAS draws on a naturally occurring phenomenon called auditory-motor entrainment. During entrainment, an external auditory rhythm enables subconscious synchronization between the auditory and motor systems to drive coordinated movement patterns [10,11]. RAS has shown clinical benefits related to walking for patients with stroke across the subacute and chronic phases in many studies, several of which are randomized controlled trials (RCTs) [12–18]. In particular, speed, step length, cadence, balance, and dynamic postural stability [12,19,20] have been shown to improve in people who have had a stroke and receive RAS. In addition, the US Department of Veterans Affairs incorporated rhythmic auditory cueing into its clinical practice guidelines for the management of stroke rehabilitation in 2019 [21].
Currently, clinicians administer the RAS protocol in rehabilitation hospitals or clinics, while accessible at-home RAS-based interventions are nonexistent. Rehabilitation at home can carry benefits including half the risk of readmission, lower caregiver strain [22], reduced cost, and greater patient satisfaction [23] relative to hospital rehabilitation. For those in the chronic phase of stroke recovery, physical therapy and to a greater extent RAS can be difficult to access due to limited insurance coverage and the limited number of neurologic music therapists who deliver RAS. The lack of a solution for at-home walking rehabilitation is a critical gap in chronic stroke recovery, and it is imperative that solutions that are safe and effective to use are developed and delivered to address this unmet need.
To help close this gap, MedRhythms has designed MR-001 (InTandem, MedRhythms, Inc), a neurorehabilitation system that delivers a RAS-based intervention for chronic stroke walking impairment and is intended to be used independently at home. […]

[Abstract] Functional MRI Assessment of Brain Activity During Hand Rehabilitation with an MR-Compatible Soft Glove in Chronic Stroke Patients: A Preliminary Study
Posted by Kostas Pantremenos in Neuroplasticity, Paretic Hand, Radiology/Imaging technology, Rehabilitation robotics on November 17, 2023
Abstract:
Brain plasticity plays a significant role in functional recovery after stroke, but the specific benefits of hand rehabilitation robot therapy remain unclear. Evaluating the specific effects of hand rehabilitation robot therapy is crucial in understanding how it impacts brain activity and its relationship to rehabilitation outcomes. This study aimed to investigate the brain activity pattern during hand rehabilitation exercise using functional magnetic resonance imaging (fMRI), and to compare it before and after 3-week hand rehabilitation robot training. To evaluate it, an fMRI experimental environment was constructed to facilitate the same hand posture used in rehabilitation robot therapy. Two stroke survivors participated and the conjunction analysis results from fMRI scans showed that patient 1 exhibited a significant improvement in activation profile after hand rehabilitation robot training, indicative of improved motor function in the bilateral motor cortex. However, activation profile of patient 2 exhibited a slight decrease, potentially due to habituation to the rehabilitation task. Clinical results supported these findings, with patient 1 experiencing a greater increase in FMA score than patient 2. These results suggest that hand rehabilitation robot therapy can induce different brain activity patterns in stroke survivors, which may be linked to patient-specific training outcomes. Further studies with larger sample sizes are necessary to confirm these findings.
[BLOG POST] Robotic neurorehabilitation for the upper limb – new insights
Posted by Kostas Pantremenos in Neuroplasticity, Paretic Hand, Rehabilitation robotics on November 13, 2023

Authors: Mihaela Molnar, Oana Vanta
1 Introduction | Robotic neurorehabilitation for the upper limb
2 The importance of robotic neurorehabilitation
4 A brief introduction to wristbot
5 Latest advances in neurorehabilitation involving robotic devices
6 Multidisciplinary evaluation of recovery – assessment of sensorimotor performance
7 The promises of robotic devices in neurorehabilitation
Introduction | Robotic neurorehabilitation for the upper limb
Chronic and incapacitating neurological conditions significantly burden families and society [1]. Brain injuries and other neurological pathologies can negatively impact a patient’s life quality by causing motor or sensory loss or dysfunction [2]. One of the leading causes of mortality and disability around the globe is stroke [3]. After a stroke, patients frequently have sensorimotor impairments, the upper limb is affected in more than two-thirds of affected individuals, and half of them experience a persisting loss of arm function [2].
Spasticity in the upper extremities affects 17% to 40% of stroke survivors, making it harder for them to perform activities of daily living (ADL). Upper-limb therapy is essential in the first six months following the stroke because, after that time, stroke survivors’ motor and ADL recovery diminishes. After six months following a stroke, 33% to 66% of individuals do not regain functional use of their upper extremities [3].
The importance of robotic neurorehabilitation
Innovative robotic devices have been created over the past few decades to assist physicians in neurorehabilitation [2]. Traditional post-stroke rehabilitation methods include:
- “hands-on” therapy (manual therapy techniques)
- constraint-induced movement therapy
- repetitive task training
- mirror therapy.
They frequently call for patients to manually perform a partial or full-assisted movement in arm/hand joints while being observed by therapists.
Robot-assisted treatment
The cost-effectiveness of traditional therapeutics has been constrained by how labor- and time-intensive they are. Robotic devices are used in robot-assisted treatment (RT), a revolutionary post-stroke rehabilitation strategy, to give patients motor or task-oriented training. Stroke survivors can perform independent training with less supervision from therapists, receive timely feedback on their performance from robotic devices, and achieve better adherence to treatment with the introduction of games or interactive upper-limb tasks, in addition to providing repetitive and high-intensity training in a cost-effective manner [3].
Robotic neurorehabilitation is appealing due to its ease of deployment potential, adaptability to various motor impairments, and excellent measurement reliability [4]. One of the major objectives of rehabilitation for people with neurological diseases is to increase mobility. High dosage and intensity, sufficient practice, specific objectives, motivation, and specialized expertise are all crucial for improving outcomes in neurorehabilitation [1].
Robotic-assisted gait training (RAGT), which may deliver a stronger treatment dosage than standard rehabilitation, is anticipated to increase mobility more successfully than conventional therapy [1]. In this application sector, “robotic technology” refers to any mechatronic device with a certain level of intelligence that may physically influence the patient’s behavior to optimize and improve his or her sensorimotor rehabilitation [2].
These robots have two main functions:
- Evaluate human sensorimotor function
- Retrain the human brain to enhance the quality of life [2].
Therefore, based on the low-level control method and every patient’s remaining abilities, each robotic device provides a pre-defined training mode. Both passive training (robot-driven, position control technique), where the robot imposes the trajectories, and active training (patient-driven), where the robot modifies its trajectory in response to the subject’s will to move, are often implemented by rehabilitation devices.
Riccardo Iandolo et al. indicate the fact that the supportive training modality, however, is the most pertinent of all the other training methods. In the active assistive training mode, the assistive controllers, which mimic the traditional physical and occupational therapy approach, aid participants in moving their impaired limbs into the proper positions when grabbing, reaching, or walking [2].
Particularly, the assistance-as-needed approach is one of the assistive techniques frequently used since it lowers the chance that the patient would only rely on the robot to complete the rehabilitation job.
On the other hand, Riccardo Iandolo et al. point to the fact that over-assistance may, in fact, reduce engagement and, hence, the potential for inducing neuroplastic alterations [2]. The “Slacking” effect, for example, develops when a patient undergoes repeated passive limb mobilization, which is defined as a decline in voluntary movement control. Challenge-based controls are used to make tasks more challenging or stimulating in addition to the assistance-as-needed strategy to stop the ‘slacking’ effect [2].
The most effective use of currently available technologies or gadgets would be a major study emphasis, even if numerous studies are concentrating on developing new ones. This might be accomplished by developing effective training techniques and implementing realistic control and assessment methods [2].
In order to optimize the potential of neurorehabilitation, robotic treatment must be combined with other disciplines, including computational neuroscience, motor learning and control, and bio-signal processing, among others [2].
ROBOTIC DEVICES USED IN UPPER LIMB MOTOR NEUROREHABILITATION
A therapy robot can be referred to as a multipurpose manipulator that can be reprogrammed and used for various rehabilitation tasks. Therefore, it would be feasible to put up a robotic system that moves the arm and hand to conduct this function, similar to locomotion robots used for gait therapy. In contrast to the lower extremity, there is no established typical motion pattern for the upper extremity, where a particular gait pattern may be identified. It requires a distinct approach due to the intricacy of the arms and hands and the wide range of motion patterns that are accessible [5].
Based on the different forms of physical human-robot interaction, there are two primary categories of robotic devices for neurorehabilitation:
- End-effector devices;
- Exoskeletons [2];
End-effector-based systems are robotic devices equipped with an exclusive interface that mechanically restrains the distal portion of the human limb, which is directly controlled; the rest of the kinematic chain is free, and the human limb has to adjust to external disturbances or movements performed by the end-effector robot [2].
Exoskeletons faithfully mimic the kinematics of the human limb and support its motion by manipulating the position and orientation of each joint. Additionally, the number of actuated joints and the range of motion (ROM) are properly selected to maximize control, resulting in close monitoring of the patient’s motion but at the cost of a higher complexity for the control of degrees of freedom (DOFs) [2].
In the past years, numerous robotic systems and protocols have been created based on task-oriented repetitive motions for the improvement of:
- ROM;
- Muscular strength;
- Movement coordination;
- Motor learning [2].
Based on the nature and degree of motor dysfunction and associated disability, one type of device may be more effective than the other; for example, exoskeletons may be more suitable to deliver forces to each joint if the patient has very low remaining sensorimotor functionality [2].
The following table exemplifies some neurorehabilitation devices (Table 1) [2,6].
| End-effector robotic devices | |
| MIT Manus | designed for the shoulder and elbow joints |
| ARM (Assisted Rehabilitation and Measurement) Guide | a counterbalanced robot that assists the reaching motion mechanically without loading the arm |
| GENTLE/s (Robotic assistance in neuro and motor rehabilitation) | |
| Italia NeReBot (Neurorehabilitation Robot) | mainly utilized to quantify aberrant joint torque coupling in chronic stroke |
| ACT (Arm Coordination Training Robot) | mainly utilized to quantify aberrant joint torque coupling in chronic stroke |
| Mirror Image Motion Enabler | implement bimanual training protocols |
| Bi-Manu-Track | implement bimanual training protocols |
| Exoskeletons devices | |
| SUEFULARMin IIICADEN (Cable-Actuated Dextrous Exoskeleton for Neurorehabilitation) RUPERT (Robotic Upper Extremity Repetitive Trainer) | Provide shoulder and elbow joint motion |
| Manovo PowerARMEO PowerIntelliArm exoskeleton | Can implement movements like hand opening and closing, grasp movements, fingers passive stretching |
Table 1. Examples of exoskeletons and end-effector devices ( available from [2,6])
In contrast to end-effector devices, upper limb exoskeletons for rehabilitation have recently been created due to the following:
- the intricate interaction between the mechanical structure of exoskeletons and the various joints in the human body,
- the intricate control schemes that must be adopted to deal with transparency and back-drivability,
- the requirement to promote patient sensorimotor recovery without passively moving the patient’s joints by using assistive training modalities capable of responding to any pathological movement [2].

Figure 1. Classification of exoskeletons and end-effector devices based on the actuation system
Several innovative control schemes for exoskeletons have recently been designed to improve inter-joint coordination [2].
End-effector and exoskeleton devices (Figure 1) implemented a closed-loop feedback control with feedforward components. This approach allows for correcting patients’ performance faults and compensation for the device mechanics’ weight, inertia, and friction. Furthermore, the robot model might generate the feedforward components or learn iteratively. This control method is typically used in exoskeletons, where position data is used to close the loop. The interaction control framework is also used to implement assistive methods. Most end-effector devices, in particular, use impedance control techniques, whereas exoskeletons use admittance control schemes [2].
The exoskeleton can function in three modes: passive (robot-driven), active (patient-driven), and challenge (robot resists applied force). A robot may also oppose the patient’s movement to make things more difficult for the patient [6]. Sliding mode controllers or controllers activated by the patient’s intention detection estimated by electrophysiological measures (e.g. surface electromyography – sEMG) and electroencephalography – EEG) have been developed in recent years for upper limb exoskeletons.
All of the approaches described above are based on comparing an error signal to a reference trajectory which may be easily estimated or created with end-effector devices; however, exoskeletons present several challenges. As previously stated, exoskeletons can restore patient inter-joint coordination by appropriately adjusting the various robot joint trajectories. Nevertheless, the relationship between recovery and exoskeleton trajectories remains unknown [2].
Recently proposed approaches include:
- Reproducing previously recorded trajectories made by healthy participants;
- Using previously recorded pathological involuntary joint torques translated in the joint kinematics domain [2].
A brief introduction to wristbot
WristBot (see Figure 2) is a robotic end-effector device intended for the neurorehabilitation of individuals who suffer neurological or orthopedic diseases allowing movements of flexion/extension in radial or ulnar deviation and pronation or supination. It has four brushless motors that allow it to guide and aid wrist motions in the three planes stated above. These motors are chosen to give precise haptic representation while compensating for the device’s weight and inertia, allowing free and smooth motions [2].
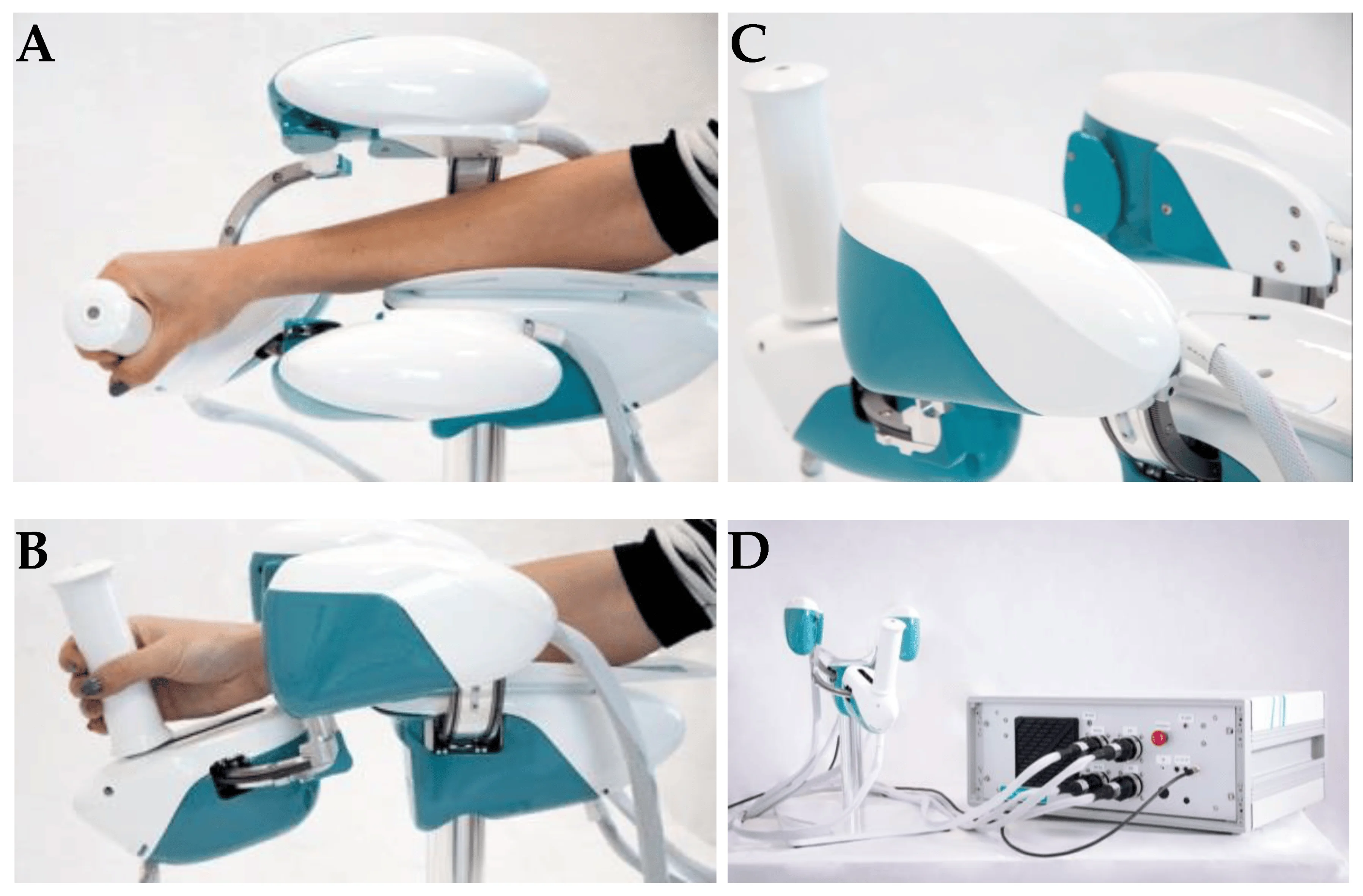
Figure 2. Wrist robot. (A) and movements in the radial-ulnar deviation DOF (degrees of freedom) (B); posterior-lateral view of WristBot’s handle (C); and frontal view of the device connected to the case, with the integrated PC and electronic control unit (D) (available from [2])
The key benefit of the WristBot is its highly customized therapy, provided by its versatility and programmability. Additionally, the device’s quantitative functional evaluation is a helpful tool to aid physicians in selecting the best course of treatment [2].
Latest advances in neurorehabilitation involving robotic devices
The neurorehabilitation process greatly benefits from robotic training. In fact, the devices may be programmed to execute several training options depending on motor learning paradigms and/or brain control. Robots can also read out information on movement performance with accuracy and precision and offer highly repeatable, exact, and reproducible movements (as dictated by forces and torques). Riccardo Iandolo et al. highlights that a suitable rehabilitation training regimen must be carefully created to make the most use of rehabilitation robots for patient care. To do this, a tremendous amount of research has been undertaken in recent years to fully understand the many mechanisms by which people (re)acquire (or relearn) a motor skill [2].
Planning an effective robot-based treatment to promote sensorimotor rehabilitation requires a thorough understanding of how the brain governs movement and which applied processes are used to learn new abilities [2].
A key factor in facilitating motor learning is motor variability. Variability was once regarded as “noise” that the brain needed to eliminate when learning, but recent research has begun to recognize its significance in developing motor abilities [2].
The benefits of receiving visual feedback from seeing someone else do an action have been amply established to enhance motor learning. According to recent research, respondents learn better when they see a video of someone executing reaching actions in a complex setting before performing the activity on their own. Motor and sensory learning are tightly connected, in the same manner, that motor learning influences sensory networks and sensory learning alters motor regions [2].
BCIs (Brain Computer Interfaces) were first designed as non-invasive communication aids, but their intrusive counterpart, commonly referred to as brain-machine interfaces (BMIs), seek to restore some degree of motor function in patients who were entirely paralyzed or severely disabled. Recent studies demonstrate how BCIs and BMIs may be effectively used to improve the results of a neurorehabilitation intervention: these systems advocate for help that mirrors the user’s motor intention, leading to greater cortical modifications.
The findings open the door to innovative, tailored therapies where closed-loop decoding of brain activity is crucial for enhancing sensorimotor repair [2].
Multidisciplinary evaluation of recovery – assessment of sensorimotor performance
The current clinical approach for assessing movement disorders mainly comprises qualitative assessments made by human operators using clinical scales, as shown in Table 2 [2].
| CLINICAL RATING SCALES | |
| Quality of Upper Extremity Skills Test (QUEST) | Assesses movement patterns and hand function |
| Modified Ashworth Scale (MAS) | Assesses the spasticity of the upper limb |
| Fugl–Meyer Assessment (FMA) | Evaluates sensorimotor impairments |
| Melbourne Assessment of Unilateral Upper Limb Function (MAUULF) | Evaluates the quality of the movements |
| Box and Block Test (BBT) | Evaluates gross manual dexterity |
Table 2. Clinical rating scales
The following tests evaluated position sense acuity, detection of passive motion, and kinesthesia: Nottinghan Sensory Assessment, Rivermead Assessment of Somatosensory Performance, and Joint Position Matching test [2].
Sensorimotor performance can also be measured via brain and/or muscle activity recordings such as high-density electroencephalography (hdEEG) recordings, source imaging methods that have lately enabled the accurate reconstruction of resting-state networks in the brain, and assessing of electrophysiological sub-cortical activity [2].
Functional connectivity (FC) represents the relationship between separate brain areas and changes after proprioceptive training with a robotic device. Moreover, FC predicts the behavioral outcomes of rehabilitation protocols and motor function recovery in stroke patients and corresponds with the extent of clinical impairment in patients with early relapsing-remitting multiple sclerosis [2].
The promises of robotic devices in neurorehabilitation
Because motor impairment is the common denominator of all functional motor disorders, focusing rehabilitation efforts on the restoration of impairment rather than compensation could be more beneficial in the acute and subacute stages of recovery. High intensity (i.e., dose per unit time), high dosage, and realistic movement training in these three dimensions are important factors influencing the impact of rehabilitation [4].
Robotic neurorehabilitation equipment may increase the quality of the quantitative evaluation, which is also necessary to comprehend better and infer the rehabilitation therapy’s effect on sensorimotor function. Robotic measures have the potential to exceed human-administered clinical scales and are only limited by the robot sensors’ performance [2]. Robots provide a more exact evaluation of both initial impairment and impairment changes in response to therapy regarding movement kinematics and dynamics [4].
Conclusion
Future research should concentrate on creating novel mechatronic structures and improved control algorithms. Exoskeletons must cope with a high level of mechanical complexity: they must be portable, lightweight, efficient, and compliant while also properly supporting the patient, even in the presence of severe impairments. In order to control costs throughout the design of a device, it is essential to keep in mind the specific disability the device is being developed to choose just the most crucial developmental goals [2].
Through the advancement of robotic scales and the incorporation of measurements with biosignals, sensorimotor performance, and recovery may be comprehensively evaluated. Researchers propose that new methods based on multimodal evaluation methodologies should be used to leverage unique and/or composite indications. This may result from understanding how to provide the greatest tools to improve motor recovery and neuroplasticity [2].
The study of Riccardo Iandolo et al. [2] highlights the importance of patient evaluation regarding rehabilitation intervention to increase its effectiveness. When building a tool to help a certain disorder, it is critical to consider the end perspective, as the user’s collaborative effort would almost certainly result in a proper treatment [2].
Robots provide movement controllability and measurement reliability, making them excellent tools for neurologists and therapists in addressing neurorehabilitation issues [4]. According to studies, robot-assisted treatment is less expensive than standard intense arm training with comparable effects. Therefore, robots are a significant aspect of the current therapeutic paradigm for improving the overall quality of neurologic rehabilitation [5].
For more information about neurorehabilitation, visit:
- Efficacy of technology-assisted gait rehabilitation in Parkinson’s disease
- Efficacy of placebo in managing pain for neurological disorders
- Neurorehabilitation in dystonia – a holistic approach
We kindly invite you to browse our Interview category: https://efnr.org/category/interviews/. You will find informative discussions with renowned specialists in the field of neurorehabilitation.
References
- Kuo CY, Liu CW, Lai CH, Kang JH, et al. Prediction of robotic neurorehabilitation functional ambulatory outcome in patients with neurological disorders. Journal of NeuroEngineering and Rehabilitation. 2021. doi:10.1186/s12984-021-00965-6
- Iandolo R, Marini F, Semprini M, Laffranchi M, et al. Perspectives and Challenges in Robotic Neurorehabilitation. Applied Sciences 2019. doi:10.3390/app9153183
- Chien WT, Chong Y, Tse MK, Chien CW, Cheng HC. Robot-assisted therapy for upper-limb rehabilitation in subacute stroke patients: A systematic review and meta-analysis. Brain and Behavior 10, 2020. doi:10.1002/brb3.1742
- Huang VS, Krakauer JW. Robotic neurorehabilitation: a computational motor learning perspective. Journal of NeuroEngineering and Rehabilitation 6, 2009. doi:10.1186/1743-0003-6-5
- Jakob I, Kollreider A, Germanotta M, Benetti F, et al. Robotic and Sensor Technology for Upper Limb Rehabilitation. Innovations Influencing Physical Medicine and Rehabilitation, 2018. doi:10.1016/j.pmrj.2018.07.011
- Rehmat N, Zuo J, Meng W, Liu Q, et al. Upper limb rehabilitation using robotic exoskeleton systems: A systematic review. International Journal of Intelligent Robotics and Applications 2, 2018, 283–295. doi:10.1007/s41315-018-0064-8
- Dehem S, Gilliaux M, Stoquart G, Detrembleur C, et al. Effectiveness of upper-limb robotic-assisted therapy in the early rehabilitation phase after stroke: A single-blind, randomised, controlled trial. Annals of Physical and Rehabilitation Medicine 62, 2019, 313-320. doi:10.1016/j.rehab.2019.04.002
[WEB] What should a stroke survivor know about the recovery plateau?
Posted by Kostas Pantremenos in Neuroplasticity, Recovery Plateau on October 29, 2023

Zachary Smith, MS, CCC-SLP |
So, you feel stuck…
Whether you’re a brain injury or stroke survivor, caregiver for someone with neurological damage, or a healthcare worker, you’ve probably heard the term “recovery plateau” when talking about the recovery process. Right after a neurological event, things can be very challenging, and the first few months afterward are when you’ll typically see the most rapid progress. During this time, people often dive into intense, acute rehab to make the most of this window and try to improve their speech, mobility, and dexterity, as much as possible.
But here’s the twist: around six months after the initial injury, stroke survivors might start hearing the term “plateau” tossed around. You might be wondering, “What exactly does that mean?” If so, don’t stress—this post is here to demystify the concept of the neurological recovery plateau and give you the information you need to take your next steps forward with confidence!
What is the neurological recovery plateau?
When we talk about stroke recovery, “plateau” is often used to describe a phase when progress starts to flatten out or slow down. Right after a neurological event, recovery can be quite rapid. But at some point, stroke survivors can feel like things aren’t getting better as quickly. During this time, you might find yourself working on the same skills in therapy sessions over and over again. Your therapist might even mention the possibility of ending therapy, leading you to wonder: “Where do I go from here?”
Will stroke survivors be stuck in a plateau forever?
Stroke survivors, we hear you: Recovery plateaus are no fun. If it feels like you’re not progressing, you might start to think that where you are now is where you will always be. But is that really true?
The short answer is: No!
What does the research say about stroke survivors and recovery plateaus?
At BrainWire, we’re constantly in awe of the brain’s resilience and complexity—and recovery plateaus are no exception! Even if it’s been a while since your neurological event, there’s solid evidence that shows you can keep making progress! A study by Basso & Macis (2011) found that people with chronic aphasia can still make neurological process through an intensive program, even after being previously discharged from speech therapy. Likewise, research from Santhanam et al (2018) found that people were able to benefit from the effects of neuroplasticity even if their stroke had occurred more than a decade prior! To cap it off, Moss & Nicholas (2006) found that the amount of time that’s passed since a stroke does not determine how much therapy can help you.
So, when you’re feeling disheartened, remember: Your brain can find ways to rewire itself to get better, even if it’s been a while since your neurological injury!
What steps can I take if I’ve met a recovery plateau?
If you’re in a recovery plateau and you’ve been wondering, “Should I keep going with or restart therapy?”, the answer is a resounding yes! The studies mentioned above are just a taste of the research within the field that shows that therapy can make an impact even years after your neurological event. In other words, that idea of “this is where I’ll always be stuck” doesn’t really apply.
Recovery isn’t a one-size-fits-all journey, and each stroke survivor takes a different route to reach their goals. While traditional speech therapy can be effective, sometimes clinicians working within healthcare institutions have their hands tied due to insurance restrictions. While the science is clear that there is no linear path to recovery, insurance companies often require evidence of rapid and steady progress to continue covering services.
But your recovery journey doesn’t have to be stymied if you find yourself in that situation! Constant Therapy can step in to help you continue striving toward your speech, language, and cognitive goals. With Constant Therapy, stroke survivors have an entire library of evidence-based exercises at their fingertips, and you can move through them at your own pace! This means that you can keep getting therapy right from your home, and you move from “I are where I am” to “I am working towards where I want to be!”
Give Constant Therapy a try for free. Click here to start your 14-day trial today.
References
Basso, A. & Macis, M. (2011). Therapy efficacy in chronic aphasia. Behavioral Neurology, 24, 317-325.
Moss, A. & Nicholas, M. (2006). Language rehabilitation in chronic aphasia and time postonset: A review of single-subject data. Stroke, 37, 3043-3051.
Santhanam, P., Duncan, E.S., & Small, S.L. (2018). Therapy-induced plasticity in chronic aphasia is associated with behavioral improvement and time since stroke. Brain Connect, 8(3), 179-188.
-
You are currently browsing the archives for the Neuroplasticity category.
Neuroplasticity


